In our previous blog post, we explored some of the most commonly used structures for expression and purification of your protein binding partners. The expression systems we addressed included bacterial or eukaryotic cell expression vehicles, which come with distinct pros and cons, depending on the properties or intended future usage of your protein(s). Moreover, we also touched on how to setup an adequate purification scheme based on the intended usage of your protein(s). We described how size exclusion and affinity chromatography can provide a rapid and well-rounded regime to secure highly purified proteins for downstream assays.
In this article, we will discuss a variety of techniques employed for identifying potential protein binding partners and show how you can use benchtop surface plasmon resonance to get the binding kinetics data needed to publish faster.
Identification of Protein Binding Partners
Protein-protein interactions are important for biological processes at both cellular and systems levels. These processes include gene expression, cell growth, cell cycle control, morphology, motility, intercellular communication and apoptosis. Studying how proteins interact with each other and identifying biological networks is vital to understanding how proteins function within the cell, and the majority of researchers will rely on this type of analysis at some point in their research.
However before biomolecular interaction analysis can take place, we must first identify potential interacting partners; how do we do this and what factors should we take into consideration?
Co-immunoprecipitations (Co-IPs) provide a low-cost method to identify interacting protein partners. An antibody specific to your previously purified binding partner of interest will “pull down” your protein and any proteins and/or ligands bound to it. In this respect, it is possible to identify known and unknown interacting partners. However, it should be noted that a “pull-down assay” is actually a separate term for a Co-IP in which an antibody for the bait protein is not available and tagging (ie. polyHis or biotin) can be used instead; CoIPs and pull down assays essentially take the affinity chromatography technique described in our previous article, one step further. Notably, Co-IPs are often used in tandem with mass spectroscopy which provides a rapid, sensitive, and reliable structure to identify interactors. Although Co-IPs and pull-downs will provide qualitative “yes or no” binding data, these will not provide true insight into what’s really taking place during an interaction event and the experimental process can be quite tedious. In this respect, it is important to incorporate quantitative techniques for a deeper understanding of the binding events. It should also be noted that nonspecific interactions between the employed antibody or bait protein can result in large numbers of false positives and this trend is observed in many protein interaction screening systems, such as yeast two-hybrid and covalent cross-linking.
Native electrophoresis and radiolabeling has emerged as a promising, sensitive and easily validated technique to identify unknown binding partners. Essentially, a protein of interest is expressed with a radiolabel, commonly [35S]Met, in a cell-free expression system, purified and allowed to incubate with cell extract to interact with its partners; notably, this is why it’s important to take into account what downstream assays you may undertake so you can select the appropriate expression and purification systems early on! Subsequently, the radiolabeled protein and cell extract is run through native polyacrylamide gel electrophoresis; importantly, native PAGE will allow all interacting partners to remain in their native conformation and allows all associations to be preserved. Bands are then exposed to an X-ray film which allows for rapid and unambiguous positive identification of the radiolabeled protein and interacting partners. Positively identified bands can then be excised from the native gel and similar to CoIPs and pull-down assays, mass spectroscopy should be employed to identify the interacting partners. This technique can save time, costs, increase reproducibility and reduce the amount of false positives. However, it should be noted that since radiolabeling is required, it can be quite hazardous to the technician and label-free techniques are typically more advantageous.
Kinetic Characterization of Interacting Partners
Upon thorough understanding and identification of interacting protein partners through CoIPs, Y2H, radiolabeling or mass spectrometry, researchers can continue on to characterize these interactions in a much more quantitative way. The techniques we’ve described for identification of binding partners above are quite standard methods and will provide insight into what is interacting, but in order to stay ahead of reviewers, it’s vital to incorporate more comprehensive and quantitative technologies to show how things are interacting. With that said, there are a few techniques that can provide quantitative binding data including ITC, MST and BLI as well as fluorescent techniques. However, these technologies typically use a lot of your sample, require labels, do not provide enough information and/or require large capital expenditure.
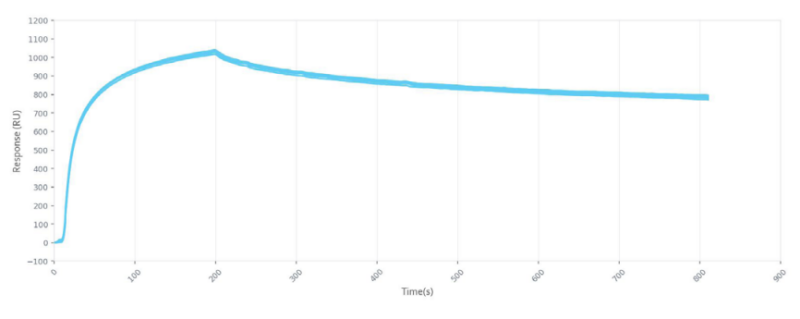
Notably, Surface Plasmon Resonance (SPR) has been the gold standard for obtaining quantitative binding kinetics data for a number of decades. As a technique, SPR is label-free, does not require a lot of sample, provides quantitative binding data and does not require hundreds of thousands of dollars in capital. Interestingly enough, SPR can actually be applied as a dual-use technology; SPR can be used in the previous workflow step of identification of binding partners in tandem with MS, known as “ligand fishing”.
Since we’ve expressed and purified at least one of our binding partners in previous steps, let’s assume that we have a purified, biotinylated extracellular domain of a receptor and we’d like to identify binding partners with the SPR-MS technique. We would select a streptavidin sensor chip and immobilize this biotinylated receptor on the sensor. From here, we can take a cell lysate which contains the potential interacting protein partners and inject this into the SPR instrument. Immediately, you would observe a large response, indicating that many interactors have bound the protein bait and at this point, all running buffer flow should be stopped to avoid losing interactors with fast off-rates. Subsequently, you would uninstall the sensor from the instrument (which has your protein bait and interactors on the surface of the sensor) and place the sensor in a non-destructive elution buffer to remove all prey protein partners and prepare samples for MS for identification. Notably, ligand fishing experiments should be conducted repeatedly so that adequate sample quantities can be extracted for MS. Using SPR-MS to identify binding partners allows for increased streamlining, throughput, reproducibility and a reduction of false positives; this is all due to the fact that a continuous flow system is used and more parameters to eliminate false positives can be optimized.
After you’ve identified your protein binding partners with SPR, you can move on to determining the kon, koff, and KD of interactions, label-free, gaining deeper insight into binding events compared to other techniques that only give endpoint measurements, such as pull-down assays. SPR is necessary not only for publications but for the advancement of many fields of medicine and medical research as can be seen below with the significant increase in publications that rely on SPR data.
Importantly, OpenSPR is a user-friendly and low maintenance benchtop SPR solution that is currently being used by hundreds of researchers. With access to SPR technology on your own lab bench, you can get the high-quality data you need to accelerate your research and publish faster.